The projects that the group members work on can be placed into seven main categories.
Biological systems are complex and the observed variablity between samples may be explained by different kinds of 'omic' information, such as the transcriptome or the proteome. However, different omics varieties exhibit unique characteristics and require tailored approaches to draw meaning from these datasets by addressing statistical issues such as sample mismatch, missing values, imbalance of features and differing requirements regarding normalisation or standardisation. Our research focuses on building better predictive models for biomarker discovery, which involves developing methods to examine clinical-pathological, mutation and omics data for prognostic biomarker discover, for example constructing suitable weights to improve weighted lasso.
Liu, C., Huang, H. and Yang, P. (2020) Multi-task learning from multimodal single-cell omics with Matilda. Nucleic Acids Research. 51(8):e45.
Jayawardana, K., Schramm ,S.J., Haydu, L., Thompson, J.F., Scolyer, R.A., Mann G.J., Müller S. and Yang J.Y. (2015) Determination of prognosis in metastatic melanoma through integration of clinico-pathologic, mutation, mRNA, microRNA, and protein information. International Journal of Cancer. 136(4):863-874.
Strbenac, D., Mann, G., Ormerod, J. and Yang, J. (2015) ClassifyR: an R package for performance assessment of classification with applications to transcriptomics. Bioinformatics. 31(11):1851-1853.
Patrick, E., Buckley, M., Müller, S., Lin, D. and Yang, J. (2015) Inferring data-specific micro-RNA function through the joint ranking of micro-RNA and pathways from matched micro-RNA and gene expression data. Bioinformatics. 31(17):2822-2828.
There are currently 3 positions open to applicants.
Biological systems are made up of a highly connected set of features and often feature built-in redundancy, so that damage to one component may be unimportant if another component which can perform the same function is undamaged. Considering biological molecules in context of their upstream regulators and downstream targets, as well as alternative pathways that lead to the same target allows better association between measured biological changes and observed phenotypes. Statistical models need to incorprate uncertainty into the inference procedure because network models are obtained from publicly available databases derived from datasets comprised of different organisms and different cell types than the ones being studied.
Ghazanfar, S. and Yang, J. Y. (2016) Characterizing mutation-expression network relationships in multiple cancers. Computational Biology and Chemistry. 63:73-82.
Wang, R., Jiang, K., Feldman, L. J., Bickel, P. J. and Huang, H. (2015) Inferring gene-gene interactions and functional modules using sparse canonical correlation analysis. Annals of Applied Statistics. 9(1):300-323.
Barter, R., Schramm, S., Mann, G. and Yang, J. (2014) Network-based biomarkers enhance classical approaches to prognostic gene expression signatures. BMC Systems Biology. 8(Supplement 4):S5.
Kim, H., Osteil, P., Humphrey, S., Cinghu, S., Oldfield, A., Patrick, E. Wilkie, E., Peng, G., Suo, S., Jothi, R., Tam, P. and Yang, P. (2020) Transcriptional network dynamics during the progression of pluripotency revealed by integrative statistical learning. Nucleic Acids Research. 48(4):1828-1842.
There are currently 2 positions open to applicants.
Methods are being developed to bring together information from health or phenotype data together with omics information to provide better predictions of patient responses and outcomes. Apart from data integration, the understanding of signalling, proteomic and transcriptional regulation is a focus of research, in particular for stem cell differentiation and early development. Such understanding is crucial for precision medicine as it builds the foundation for testing drugs, treating or preventing birth defects, as well as other complex diseases caused by the malfunction of the complex regulatory networks.
Patrick, E., Schramm, S.-J., Ormerod, J.T., Scolyer, R.A., Mann, G. J., Müller, S. and Yang J. Y. (2017) A multi-step classifier addressing cohort heterogeneity improves performance of prognostic biomarkers in three cancer types. Oncotarget. 8:2807-2815.
There is currently 1 position open to applicants.
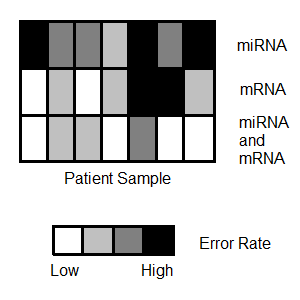
Many classification tasks with high-throughput biological data have high error rates, suggesting multiple distinct features classify different subsets of individuals or that simple changes do not capture the full set of differences present among classes (e.g. recovery vs. cancer relapse). Motivated by complex data, including for longitudinal, clustered and correlated univariate and multivariate responses, classification and improved prediction methods are developed for multi-omics data. Theoretical properties of regularization methods in various asymptotic scenarios are investigated and improvements to these methods have been made by learning from resampling-based stability information. Advanced model visualisation methods are implemented to enable interactive and dynamic model building allowing for the investigation of robust selection and estimation methods.
Tarr, G., Müller, S. and Welsh, A. (2017) mplot: An R package for graphical model stability and variable selection. Journal of Statistical Software. To appear.
Strbenac, D., Mann, G. J., Yang, J. Y. and Ormerod, J.T. (2017) Differential distribution improves gene selection stability and has competitive classification performance for patient survival. Nucleic Acids Research. 13:e119.
There is currently 1 position open to applicants.
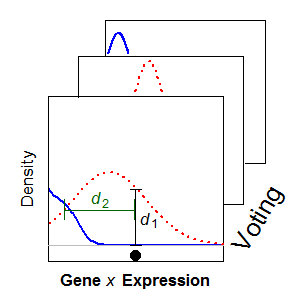
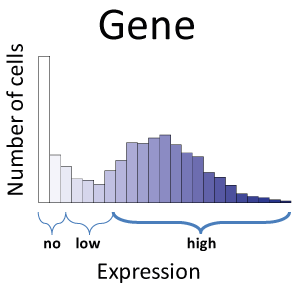
Until recently, heterogeneity across single cells in a biological tissue extract had been lost when a sample comprising thousands of cells was measured on the same assay, providing only an average measurement and thus discarding information about cell-to-cell variation. Our research focuses on analysis of single cell RNA-Seq (scRNA-Seq) data, but extends to other technology, such as CyTOF. We have employed statistical mixture modelling to reduce the impact of noisy measurements and to characterise the expression profiles of genes across populations of single cells. We have used this framework to characterise the expression profiles of single neurons using a variety of publicly available scRNA-Seq datasets.
Kim, H.J., Wang, K., Chen, C., Lin, Y., Tam, PPL., Lin, D.M., Yang, J.Y.H. and Yang, P. (2021) Uncovering cell identity through differential stability with Cepo. Nature Computational Science. 1:784-790.
Lin, Y., Cao, Y., Kim, H.J., Salim, A., Speed, T., Lin, D.M., Yang, P. and Yang, J.Y.H. (2020) scClassify: sample size estimation and multiscale classification of cells using single and multiple reference. Molecular Systems Biology. https://doi.org/10.15252/msb.20199389
Kim, H.J., Lin, Y., Geddes, T.A., Yang, J.Y.H. and Yang, P. (2020) CiteFuse enables multi-modal analysis of CITE-seq data. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa282
Lin, Y., Ghazanfar, S., Wang, K.Y., Gagnon-Bartsch, J.A., Lo, K.K., Su, X., Han, Z.G., Ormerod, J.T., Speed, T.P., Yang, P. and Yang, J.Y.H. (2019) scMerge leverages factor analysis, stable expression, and pseudoreplication to merge multiple single-cell RNA-seq datasets. Proceedings of the National Academy of Sciences. 10.1073/pnas.1820006116
Kim, T., Chen, I., Lin, Y., Wang, A., Yang, J. and Yang, P. (2018) Impact of similarity metrics on single-cell RNA-seq data clustering. Briefings in Bioinformatics. 10.1093/bib/bby076
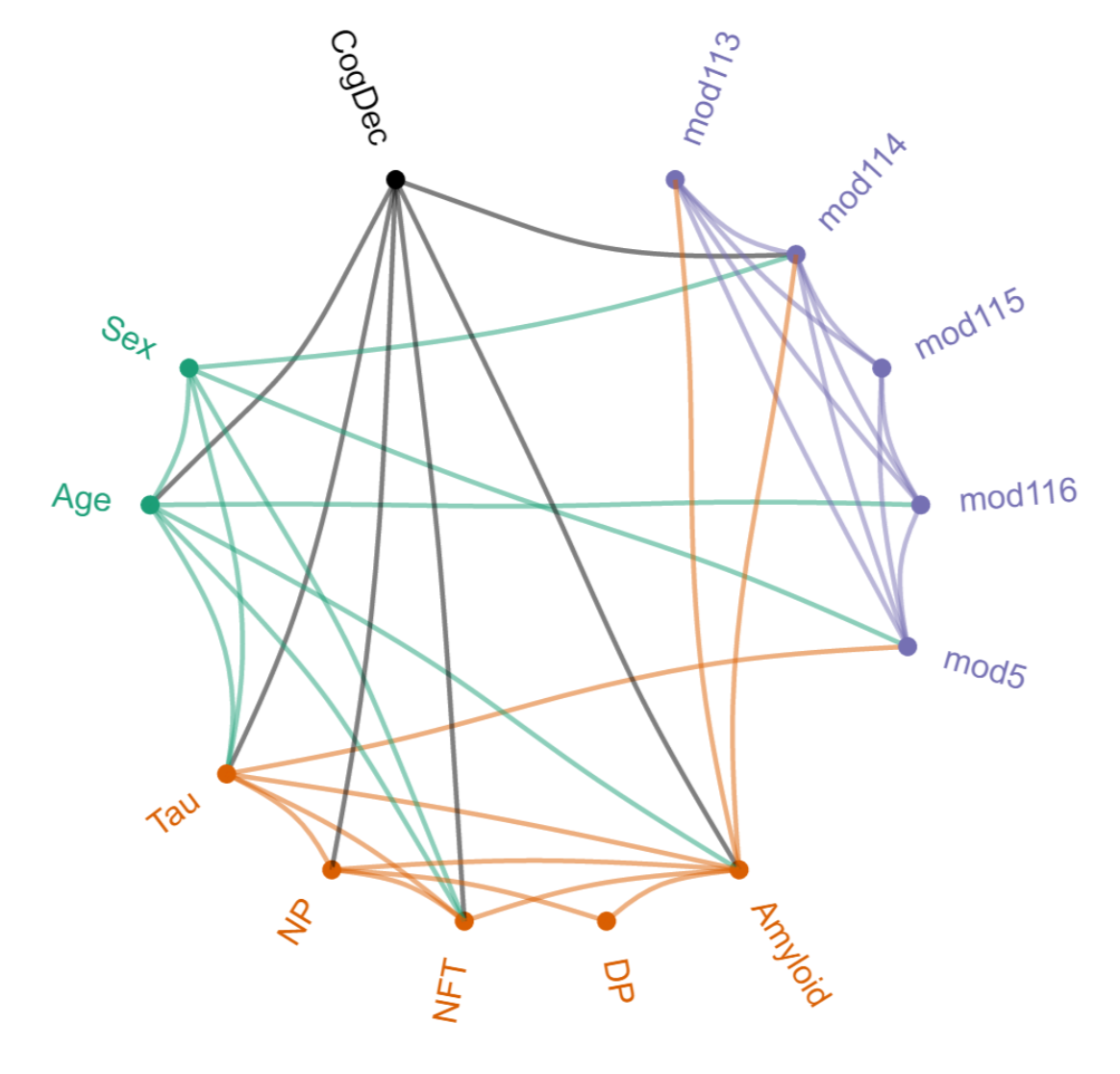
Recently, the capacity to analyse the large complex datasets generated in studies of brain function and related disorders has begun being implemented. This benefits from the team's strengths in hypothesis formulation, network construction, image analysis and data integration to extract information from various high-throughput technologies such as functional magnetic resonance imaging (fMRI) and microscopy.
Patrick E., Rajagopal, S., Wong, H-K. A., McCabe C., Xu, J., Tang, A., Imboywa, S.H., Schneider, J.A., Pochet, N., Krichevsky, A.M., Chibnik, L.B., Bennett, D.A. and de Jager, P .L. (2017) Dissecting the role of non-coding RNAs in the accumulation of amyloid and tau neuropathologies in Alzheimer's disease. Molecular Neurodegeneration. 12:51.
There is currently 3 positions open to applicants.
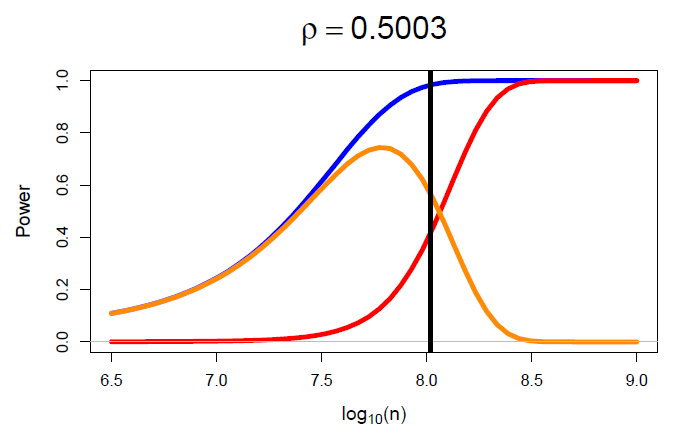
Methods in this category are sometimes necessary for large, complex problems. By exploiting the flexibility of Bayesian hierarchical modelling framework, these models can be fit using variational Bayes, expectation propagation and Laplace-like approximations in mere minutes whereas exact methods, such as Markov chain Monte Carlo (MCMC), could take days to produce a result.
With the advent of both high-throughput genotyping and phenotyping technologies, there is now an abundance of data. The optimal way to design and collect these data and the inference of meaningful interpretation from these big datasets is the focus of research efforts. Strong links with Meat and Livestock Australia have also been established.
Konarska, E., Kuchida, K., Tarr, G. and Polkinghorne, R.J. (2017). Relationships between marbling measures across principal muscles. Meat Science. 123:67-78.